GLIMPSE2 Low Pass Imputation Overview
| Pipeline Version | Date Updated | Documentation Author | Questions or Feedback |
|---|---|---|---|
| Glimpse2LowPassImputation_v0.0.10 (pre-release) | May, 2026 | Terra Scientific Pipeline Services | Please file an issue in WARP. |
Introduction to the GLIMPSE2 Low Pass Imputation pipeline
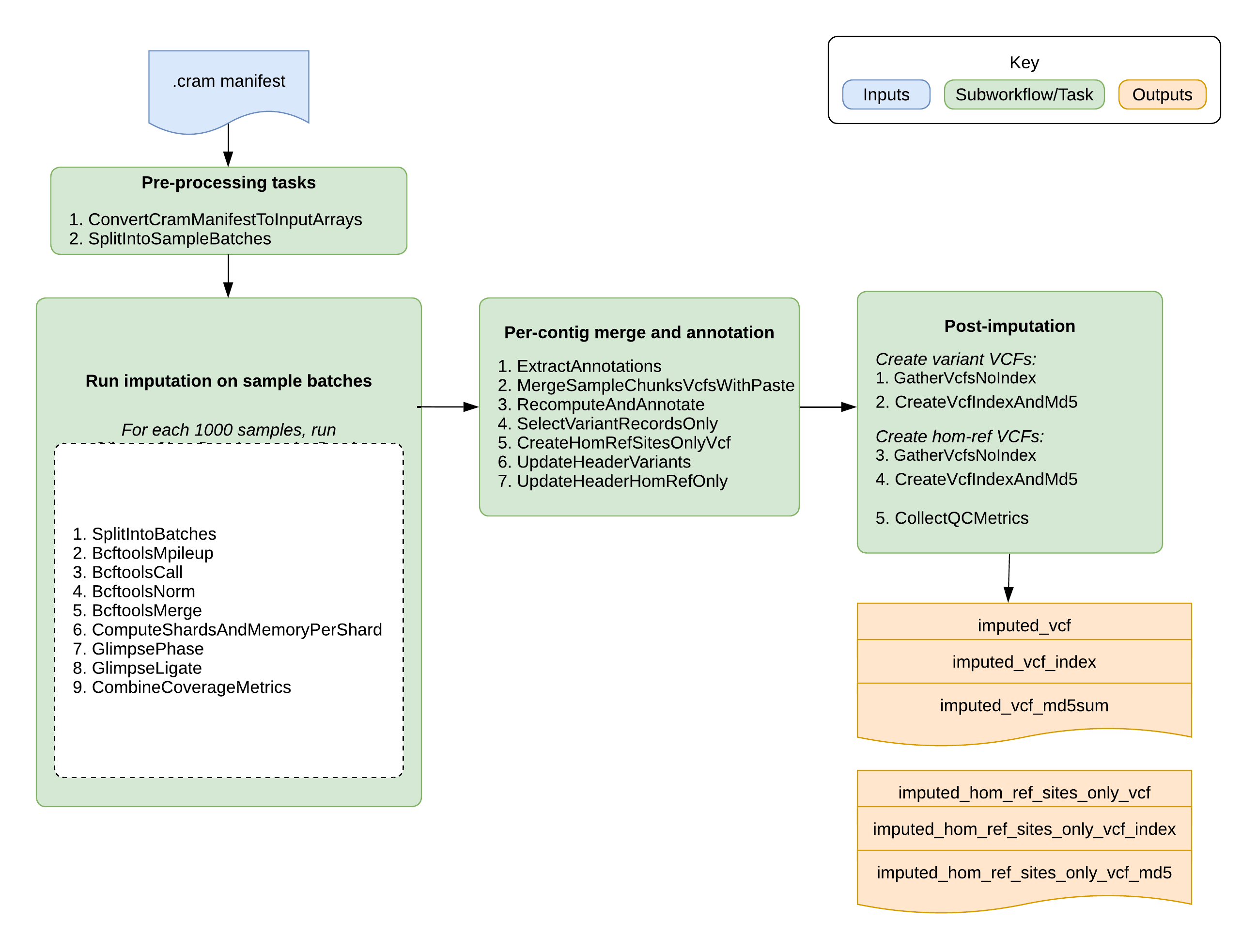
The GLIMPSE2 Low Pass Imputation pipeline imputes missing genotypes from a list of low-pass CRAM/CRAI files (or a sample manifest pointing to GCS file paths) using a large genomic reference panel. It uses GLIMPSE2 as the imputation tool. Overall, the pipeline splits samples into batches, performs variant calling and imputation on each batch across genomic chunks, and merges the results into a final multi-sample VCF. It outputs the imputed VCF along with key imputation metrics.
GLIMPSE2 Low-Pass Imputation Summary
The Glimpse2LowPassImputation workflow is a WDL-based pipeline for low-pass whole genome imputation using GLIMPSE2.
This top-level workflow is now a gateway that scales to large cohorts by splitting samples into batches, running a per-batch imputation subworkflow, then merging batch outputs back into cohort-level results.
The workflow processes each requested contig independently, imputes each sample batch against reference-defined chunks, ligates chunk outputs per batch/contig, merges sample columns across batches, recomputes AF/INFO annotations, and gathers contig outputs into final genome-wide files.
Pipeline Features
| Pipeline features | Description | Source |
|---|---|---|
| Assay type | Low-pass whole genome imputation using GLIMPSE2 | GLIMPSE2 |
| Overall workflow | CRAM calling, shard-based phasing/imputation, ligation, batch merge, and QC | Defined in Glimpse2LowPassImputation.wdl + imported subworkflows/tasks |
| Workflow language | WDL 1.0 | openWDL |
| Sub-workflows | Gateway workflow + Glimpse2LowPassImputationBatch | Imported from Glimpse2LowPassImputationBatch.wdl |
| Genomic processing | Contig-by-contig and reference-chunk-based processing | Workflow scatter logic |
| Cohort scalability | Inputs optionally specified via cram_manifest, sample batching (sample_batch_size) then per-contig batch merge | Gateway orchestration in Glimpse2LowPassImputation.wdl |
| Algorithms | bcftools mpileup/call/norm/merge + GLIMPSE2 phase/ligate + post-merge re-annotation | Task commands in batch and task WDLs |
| Quality control | Sample QC metrics and optional coverage-metrics aggregation | CollectQCMetrics, CombineCoverageMetrics |
| Data input file format | CRAM/CRAI arrays with sample IDs | Workflow input block |
| Data output file format | Imputed VCFs, indexes, md5s, and QC/coverage metric tables | Workflow outputs |
| Containers | GATK, GLIMPSE2, bcftools/samtools suite, Hail, Python, Ubuntu | Runtime blocks |
| Resource optimization | Parallelization by sample batch, contig, and reference shard | Workflow architecture |
Inputs
This gateway workflow expects CRAM-based inputs and a GLIMPSE2-compatible reference panel layout.
| Input | Description |
|---|---|
cram_manifest | Optional manifest TSV file containing columns (including header line) of sample_id, cram_path, and cram_index_path referring to cloud-hosted input files to be imputed. This or all three array inputs (crams, cram_indices, sample_ids) must be provided. |
crams | Optional array of input CRAMs |
cram_indices | Optional array of CRAI files corresponding to crams |
sample_ids | Optional array of sample ID strings corresponding to CRAM inputs |
contigs | Array of contigs/chromosomes to process |
reference_panel_prefix | Directory/prefix containing sites.<contig>.vcf.gz, sites_table.<contig>.gz, and reference_chunks.<contig>.txt |
fasta | Reference FASTA |
fasta_index | FASTA index |
output_basename | Basename for intermediate and final outputs |
ref_dict | Reference dictionary used during ligation/header normalization |
impute_reference_only_variants | Whether to impute reference-only variants (default: false) |
call_indels | Whether to include indels during calling/imputation (default: false) |
calling_batch_size | Batch size for CRAM calling inside each batch subworkflow (default: 100) |
sample_batch_size | Batch size at gateway level for splitting very large cohorts (default: 1000) |
glimpse_phase_cpu_override | Optional cpu override for GlimpsePhase task (default: 4) |
pipeline_header_line | Optional additional header lines to add to the output VCF |
gatk_docker | GATK Docker image |
glimpse_docker | GLIMPSE2 Docker image |
docker_merge | Docker used for merge/re-annotation step |
info_filter_for_inclusion | Optional minimum INFO score threshold; variants below this value are excluded from the final output VCF (default: 0.0) |
Workflow Tasks
The top-level workflow orchestrates batching, per-batch imputation, and cohort-level merging/re-annotation.
| Task / Call | Purpose | Input Dependencies | Key Function |
|---|---|---|---|
ConvertInputArraysToManifest | Convert input CRAMs/CRAIs/sample IDs arrays into cram manifest | crams, cram_indices, sample_ids | Facilitates submission of very large sets of inputs via manifest file |
SplitIntoSampleBatches | Split CRAMs/CRAIs/sample IDs in cram manifest into sample-level batches | crams, cram_indices, sample_ids from inputs or derived from cram_manifest, sample_batch_size | Enables large-cohort scaling at gateway level |
RunBatch (Glimpse2LowPassImputationBatch) | Run full low-pass imputation pipeline on each sample batch | Batch-specific CRAMs/indices/sample IDs + reference inputs | Produces per-batch, per-contig ligated imputed VCFs |
ExtractAnnotations | Extract AF/INFO annotations from each batch contig VCF | Batch ligated VCFs and indexes | Captures annotations needed for post-merge recomputation |
MergeContigVcfs (MergeSampleChunksVcfsWithPaste) | Merge sample columns across batch VCFs for one contig | Array of batch VCFs for contig | Creates full-cohort contig VCF with aligned site lists |
RecomputeAndAnnotate | Recompute AF/INFO across merged cohort and write updated contig VCF | Merged contig VCF + extracted annotations | Restores cohort-correct annotations after paste-based merge |
FilterVcfByInfo | Filter variants below the INFO score threshold (optional) | Re-aanotated VCF; only runs when info_filter_for_inclusion is supplied | Removes low-quality imputed variants from the final VCF |
SelectContigVariants | Create variants-only contig VCF | Re-annotated contig VCF | Removes homozygous-reference-only records |
UpdateHeaderVariants | Update variants-only VCF header with reference info | Filtered VCF or re-annotated VCF | Updates VCF headers with reference dictionary information and optionally adds pipeline metadata |
CreateContigHomRefVcf | Create hom-ref-sites-only contig VCF | Re-annotated contig VCF | Keeps homozygous-reference-only sites |
UpdateHeaderHomRefOnly | Update hom ref sites only VCF header with reference info | Filtered VCF or re-annotated VCF | Updates VCF headers with reference dictionary information and optionally adds pipeline metadata |
MergeBatchCoverageMetrics | Combine optional coverage metric files across batches | RunBatch.coverage_metrics | Produces aggregated coverage table when metrics exist |
GatherVcfsNoIndex | Gather contig variant VCFs into genome-wide variant VCF | Variant-only contig VCFs | Produces final genome-wide variant VCF |
CreateVcfIndexAndMd5 | Index and checksum final variant VCF | Filtered VCF (if info_filter_for_inclusion supplied) or gathered variant VCF | Creates .tbi and md5 |
GatherVcfsNoIndexHomRefOnly | Gather contig hom-ref-sites-only VCFs | Hom-ref contig VCFs | Produces final genome-wide hom-ref-sites-only VCF |
CreateVcfIndexAndMd5HomRefOnly | Index and checksum final hom-ref-sites-only VCF | Gathered hom-ref-sites-only VCF | Creates .tbi and md5 |
CollectQCMetrics | Compute sample QC metrics from final imputed variant VCF | Filtered VCF (if info_filter_for_inclusion supplied) or gathered variant VCF | Generates sample-level QC report |
Outputs
Upon successful completion, the workflow emits final genome-wide imputed outputs, corresponding index and checksum files, and QC metrics. Coverage metrics are optional.
| Output | Description |
|---|---|
imputed_vcf | Final imputed multi-sample variant VCF |
imputed_vcf_index | Index file for final imputed VCF |
imputed_vcf_md5sum | MD5 checksum for final imputed VCF |
imputed_hom_ref_sites_only_vcf | Final sites-only VCF containing homozygous-reference-only sites |
imputed_hom_ref_sites_only_vcf_index | Index file for hom-ref-sites-only VCF |
imputed_hom_ref_sites_only_vcf_md5 | MD5 checksum for hom-ref-sites-only VCF |
qc_metrics | Sample-level QC metrics table |
coverage_metrics | Optional combined coverage metrics table |
Glimpse2LowPassImputationBatch summary
The Glimpse2LowPassImputationBatch workflow is the per-batch subworkflow used by the top-level Glimpse2LowPassImputation gateway workflow.
It is designed for cohorts up to roughly 1000 samples per batch, then returns contig-level ligated imputed VCFs that can be merged across batches upstream.
Batch Workflow Role
- runs low-pass variant calling from CRAMs at reference-panel sites
- phases/imputes each contig in reference-defined chunks using GLIMPSE2
- ligates chunk-level outputs back into one imputed VCF per contig
- emits optional coverage metrics aggregated across chunks/contigs
Batch Inputs
| Input | Description |
|---|---|
contigs | Contigs/chromosomes to process |
reference_panel_prefix | Prefix containing sites.<contig>.vcf.gz, sites_table.<contig>.gz, and reference_chunks.<contig>.txt |
cram_manifest | TSV file containing paths for crams and cram indices as well as sample ids |
fasta / fasta_index | Reference FASTA and index |
output_basename | Basename for intermediate and emitted files |
impute_reference_only_variants | Pass-through option for GLIMPSE2 phase |
call_indels | Whether to include indels during calling/imputation |
calling_batch_size | Internal batch size for CRAM calling fan-out within this subworkflow |
gatk_docker / glimpse_docker | Container images for GATK and GLIMPSE2 tools |
Batch Internal Processing
| Step | Purpose |
|---|---|
SplitCramManifestIntoBatchesOfStrings | Splits cram manifest into internal calling batches |
BcftoolsMpileup | Computes pileups at panel sites per internal batch |
BcftoolsCall | Calls candidate variants from mpileup output |
BcftoolsNorm | Normalizes and indexes called variants |
BcftoolsMerge (conditional) | Merges per-internal-batch VCFs if multiple were produced |
ComputeShardsAndMemoryPerShard | Reads reference chunks and computes per-shard memory estimates |
GlimpsePhase | Runs GLIMPSE2_phase for each reference shard |
GlimpseLigate | Ligates shard outputs to one contig-level imputed VCF |
MergeBatchCoverageMetrics (conditional) | Combines optional shard/contig coverage metric files |
Batch Outputs
| Output | Description |
|---|---|
imputed_contig_ligated_vcfs | Per-contig ligated imputed VCFs for this sample batch |
imputed_contig_ligated_vcf_indices | Index files for each contig ligated VCF |
coverage_metrics | Optional batch-level combined coverage metrics table |
Glimpse2MergeBatches AF and INFO score recalculation
As part of the batch merge and re-annotation step in the top-level workflow, we recalculate AF and INFO scores across the full cohort after merging batch VCFs. This is necessary because the batch-level VCFs contain AF/INFO annotations that are only correct for the samples in that batch, so they must be recalculated for the entire input sample set. To learn more about how we do this, see the AF and INFO score recalculations on GitHub.
Glimpse2LowPassImputationQuotaConsumed summary
The QuotaConsumed workflow computes submitted sample count for service quota accounting.
Quota is derived from number of CRAM entries found in the provided CRAM manifest.
Glimpse2LowPassImputationQC summary
The InputQC workflow validates CRAM-based inputs supplied by the cram_manifest for GLIMPSE2 low-pass imputation. Checks include:
- required manifest columns are present:
sample_id,cram_path,cram_index_path - counts of CRAMs, CRAIs, and sample IDs match
- sample IDs are unique
- CRAM paths are unique
- CRAM file names end with
.cramand indices end with.crai - input paths use
gs://format and are accessible - CRAM file sizes do not exceed the configured maximum (default: 10 GB)
- CRAM files are aligned to the expected reference
- optional requester-pays validation via
billing_project_for_rp
Important notes
- Runtime parameters are optimized for Broad's Google Cloud Platform implementation.
Citing the Imputation Pipeline
If you use the GLIMPSE2 Low Pass Imputation Pipeline in your research, please consider citing our preprint:
Degatano, K., Awdeh, A., Cox III, R.S., Dingman, W., Grant, G., Khajouei, F., Kiernan, E., Konwar, K., Mathews, K.L., Palis, K., et al. Warp Analysis Research Pipelines: Cloud-optimized workflows for biological data processing and reproducible analysis. Bioinformatics 2025; btaf494. https://doi.org/10.1093/bioinformatics/btaf494
Contact us
Help us make our tools better by filing an issue in WARP; we welcome pipeline-related suggestions or questions.
Licensing
Copyright Broad Institute, 2020 | BSD-3
The workflow script is released under the WDL open source code license (BSD-3) (full license text at https://github.com/broadinstitute/warp/blob/master/LICENSE). However, please note that the programs it calls may be subject to different licenses. Users are responsible for checking that they are authorized to run all programs before running this script.