Imputation Overview
| Pipeline Version | Date Updated | Documentation Author | Questions or Feedback |
|---|---|---|---|
| ImputationBeagle_v3.0.0 | November, 2025 | Terra Scientific Pipeline Services | Please file an issue in WARP. |
Introduction to the Array Imputation pipeline
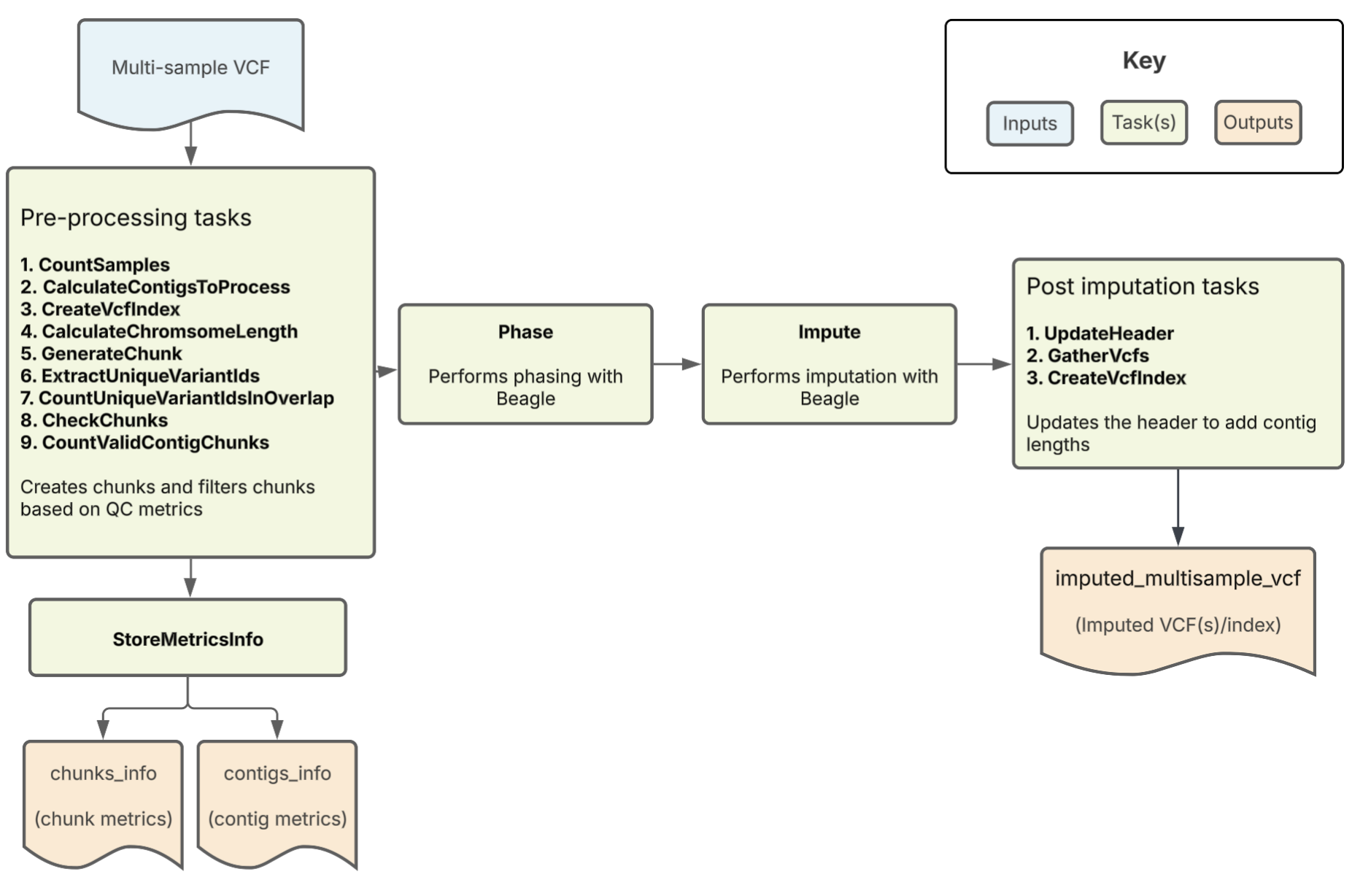
The Array Imputation pipeline imputes missing genotypes from either a multi-sample VCF or an array of single-sample VCFs using a large genomic reference panel. It uses Beagle as the imputation tool. Overall, the pipeline filters, phases, and performs imputation on a multi-sample VCF. It outputs the imputed VCF along with key imputation metrics.
Set-up
Workflow installation and requirements
The Array Imputation workflow is written in the Workflow Description Language (WDL) and can be deployed using a WDL-compatible execution engine like Cromwell, a GA4GH compliant, flexible workflow management system that supports multiple computing platforms. To identify the latest workflow version and release notes, please see the Imputation workflow changelog. The latest release of the workflow, example data, and dependencies are available from the WARP releases page. To discover and search releases, use the WARP command-line tool Wreleaser.
Using the Array Imputation pipeline This pipeline is used by the All of Us + AnVIL Imputation Service. If you choose to use this service, you can impute your samples against the 515,000+ genomes in the All of Us + AnVIL reference panel, which can provide greater accuracy at more sites.
You can alternatively run the pipeline with your own panel, using this WDL.
Input descriptions
The table below describes each of the Array Imputation pipeline inputs. The workflow requires a multi-sample VCF. These samples must be from the same species and genotyping chip.
For examples of how to specify each input in a configuration file, as well as cloud locations for different example input files, see the example input configuration file (JSON).
| Input name | Description | Type |
|---|---|---|
| multi_sample_vcf | Multi-sample VCF file containing genotype data | File |
| ref_dict | Reference dictionary for contig information and header updating | File |
| contigs | Array of allowed contigs/chromosomes to process | Array of strings |
| reference_panel_path_prefix | Path to the cloud storage containing the reference panel files for all contigs | String |
| genetic_maps_path | Path to the cloud storage containing the genetic map files for all contigs | String |
| output_basename | Basename for intermediate and output files | String |
| chunkLength | Size of chunks; default set to 25 MB | Int |
| chunkOverlaps | Padding added to the beginning and end of each chunk to reduce edge effects; default set 2 MB | Int |
| sample_chunk_size | Number of samples to chunk by when processing (default: 1,000) | Int |
| pipeline_header_line | Optional additional header lines to add to the output VCF | String |
| min_dr2_for_inclusion | Min value of DR2 to include in final output (default: 0.0) | Float |
| bref3_suffix | File extension used for the BREF3 in the reference panel (default: .bref3) | String |
| unique_variant_ids_suffix | File extension for unique variant ID files (default: .unique_variants) | String |
| gatk_docker | GATK Docker image (default: us.gcr.io/broad-gatk/gatk:4.6.0.0) | String |
| ubuntu_docker | Ubuntu Docker image (default: us.gcr.io/broad-dsde-methods/ubuntu:20.04) | String |
| error_count_override | Override for error check on chunk qc (set to 0 for workflow to continue no matter how many errors exist) | Int |
| beagle_cpu | Number of CPUs to use for Beagle Phase and Impute tasks (default: 8) | Int |
| beagle_phase_memory_in_gb | Memory, in GB, to use for Beagle Phase task (default: 40) | Int |
| beagle_impute_memory_in_gb | Memory, in GB, to use for Beagle Impute task (default: 45) | Int |
Workflow tasks and tools
The Array Imputation workflow imports a series of tasks from the ImputationTasks WDL and ImputationBeagleTasks WDL, which are hosted in the Broad tasks library. The table below describes each workflow task, including the task name, tools, relevant software and non-default parameters.
| Task name (alias) in WDL | Tool | Software | Description |
|---|---|---|---|
| CountSamples | query | bcftools | Uses the merged input VCF file to count the number of samples and output a TXT file containing the count. |
| CreateVcfIndex | index | bcftools | Creates index of input multisample vcf |
| CalculateContigsToProcess | Determine which contigs will be processed by the workflow | multi_sample_vcf, contigs | Extracts contigs from input VCF and filters by allowed contigs |
| CalculateChromsomeLength | grep | bash | Reads chromosome lengths from the reference dictionary and uses these to generate chunk intervals for the GenerateChunk task. |
| GenerateChunk | SelectVariants | GATK | Performs site filtering by selecting SNPs only and excluding InDels, removing duplicate sites from the VCF, selecting biallelic variants, excluding symbolic/mixed variants, and removing sites with a maximum fraction of samples with no-call genotypes greater than 0.1. Also subsets to only a specified chunk of the genome. |
| ExtractUniqueVariantIds | SelectVariants, query | GATK, bcftools | Extracts and counts unique variant IDs from an optionally specified interval of the input VCF in CHROM:POS:REF:ALT format |
| CountUniqueVariantIdsInOverlap | comm | bash | Counts variants in the filtered VCF chunk using unique variant ID lists; Returns the number of variants in the chunk and variants also present in the reference panel. |
| CheckChunks | convert | bcftools | Confirms that there are no chunks where less than 3 sites or less than 50% of the sites in the array are also in the reference panel; if valid, creates a new VCF output. |
| CountValidContigChunks | bash | Counts the number of valid chunks by counting true values in the validation boolean array | |
| StoreMetricsInfo | Python (pandas) | Gathers all results from CheckChunks; creates chunk-level and contig-level (chromosome-level) metrics files with variant counts | |
| ErrorWithMessageIfErrorCountNotZero | bash | Fails workflow if any chunks fail qc check. Can be overridden with error_count_override input | |
| SelectSamplesWithCut | cut | bash | Chunks vcf by sample_chunk_size if more than sample_chunk_size samples exist in input vcf |
| Phase | Beagle | Performs phasing on the filtered, validated VCF | |
| Impute | Beagle | Performs imputation on the prephased VCF; | |
| LocalizeAndSubsetVcfToRegion | SelectVariants | GATK | Remove padding from imputed vcf |
| QuerySampleChunkedVcfForReannotation | query | bcftools | Query DS, AP1, AP2 from sample chunked VCFs to be used when merging samples together |
| RemoveAPAnnotations | annotate | bcftools | Remove AP1, AP2 annotations to reduce file size now that they’re no longer needed |
| RecalculateDR2AndAFChunked | python | Used query output to summarize DS, AP1, AP2 values | |
| MergeSampleChunksVcfsWithPaste | Paste, view | Bash, bcftools | Merge sample chunked VCFs |
| IndexMergedSampleChunksVcfs | index | bcftools | Creates index for sample chunk merged VCF |
| AggregateChunkedDR2AndAF | python | Take summarized DS, AP1, AP2 data and calculate AF and DR2 | |
| ReannotateDR2AndAF | annotate | bcftools | Reannotate DR2 and AF for sample chunk merged vcf |
| FilterVcfByDR2 | Filter variants by DR2 threshold | imputed VCF, min_dr2_for_inclusion | Removes variants below provided min_dr2_for_inclusion value |
| UpdateHeader | UpdateVCFSequenceDictionary | GATK | Updates the header of the imputed VCF; adds contig lengths |
| SelectVariantRecordsOnly | Create a VCF with only variant sites | imputed VCF | Generates a VCF that only has variant sites |
| CreateHomRefSitesOnlyVcf | Create a VCF with only homozygous reference sites | imputed VCF | Generates a VCF that only has homozygous reference sites |
| GatherVcfsNoIndex | Combine all chromosome chunks, variants only | all processed chunks | Creates final output VCF |
| IndexMergedSampleChunksVcfs | Create an index for imputed output | final output VCF | Indexes are useful for downstream processing |
| GatherHomRefSitesOnlyVcfs | Combine all chromosome chunks, homozygous reference sites only | all processed chunks | Creates sites only homozygous reference output VCF |
| CreateIndexForGatheredVcfHomRefSitesOnly | Create an index for homozygous reference sites only VCF | final output VCF | Indexes are useful for downstream processing |
Workflow outputs
The table below summarizes the workflow outputs. If running the workflow on Cromwell, these outputs are found in the task execution directory.
| Output name | Description | Type |
|---|---|---|
| imputed_multi_sample_vcf | Final imputed multi-sample WGS VCF file, only contains variant records | |
| imputed_multisample_vcf_index | Index file for VCF from the CreateIndexForGatheredVcf task. | Index |
| imputed_hom_ref_sites_only_vcf | Sites only VCF that contains sites where all samples are homozygous reference | |
| imputed_hom_ref_sites_only_vcf_index | Index file for hom ref sites only VCF | |
| chunks_info | TSV from StoreMetricsInfo task; contains the chunk intervals, variant counts per chunk (filtered input and overlap with reference panel), and whether each chunk was successfully imputed. | TSV |
| contigs_info | TSV from StoreMetricsInfo task; contains contig-level (chromosome-level) aggregated metrics including total variant counts in the raw input, filtered input, and overlap with reference panel for each processed chromosome. | TSV |
Important notes
- Runtime parameters are optimized for Broad's Google Cloud Platform implementation.
Citing the Imputation Pipeline
If you use the Imputation Pipeline in your research, please consider citing our preprint:
Degatano, K., Awdeh, A., Cox III, R.S., Dingman, W., Grant, G., Khajouei, F., Kiernan, E., Konwar, K., Mathews, K.L., Palis, K., et al. Warp Analysis Research Pipelines: Cloud-optimized workflows for biological data processing and reproducible analysis. Bioinformatics 2025; btaf494. https://doi.org/10.1093/bioinformatics/btaf494
Contact us
Help us make our tools better by filing an issue in WARP; we welcome pipeline-related suggestions or questions.
Licensing
Copyright Broad Institute, 2020 | BSD-3
The workflow script is released under the WDL open source code license (BSD-3) (full license text at https://github.com/broadinstitute/warp/blob/master/LICENSE). However, please note that the programs it calls may be subject to different licenses. Users are responsible for checking that they are authorized to run all programs before running this script.