MainVcfQC
Creates plots for call set analysis and quality control (QC). This module is run by default with the FilterGenotypes module. Note, however, that the module is a stand-alone workflow and can be used with any batch- or cohort-level (i.e. multi-sample) SV VCF, including outputs from ClusterBatch and onward.
Users can optionally provide external benchmarking datasets with the site_level_comparison_datasets and sample_level_comparison_datasets parameters.
The following sections provide guidance on interpreting these plots for VCFs that have been run with the recommended pipeline through FilterGenotypes.
Output structure
The tarball output has the following directory structure:
/plots/main_plots: main QC plots, described in detail below./plots/supplementary_plots: comprehensive set of QC plots with greater detail, some of which are used as panels inmain_plots./data: various datafiles from the QC process such as family structures, a list of the samples used for plotting, variant statistics, and lists of overlapping variants against any external call sets provided.
Plots description
This section summarizes the most important plots used for overall call set QC found in the /plots/main_plots
directory.
Example plots are provided from a high-quality dataset consisting of 161 samples from the 1000 Genomes Project aligned with DRAGEN v3.7.8. Note that QC metrics and distributions will vary among data sets, particularly as sample size increases, and these examples are intended to provide a simple baseline for acceptable call set quality. Please see the Recommendations section for prescribed quality criteria.
The following plots are of variants passing all filters (i.e. with the FILTER status set to PASS or MULTIALLELIC). This is the
default behavior of the QC plots generated in FilterGenotypes.
When running MainVcfQc as a standalone workflow, users may set the
bcftools_preprocessing_options argument to limit plotted variants based on FILTER status.
For example, to limit to PASS and MULTIALLELIC variants for a VCF generated from FilterGenotypes use:
"bcftools_preprocessing_options": "-i 'FILTER=\"PASS\" || FILTER=\"MULTIALLELIC\"'"
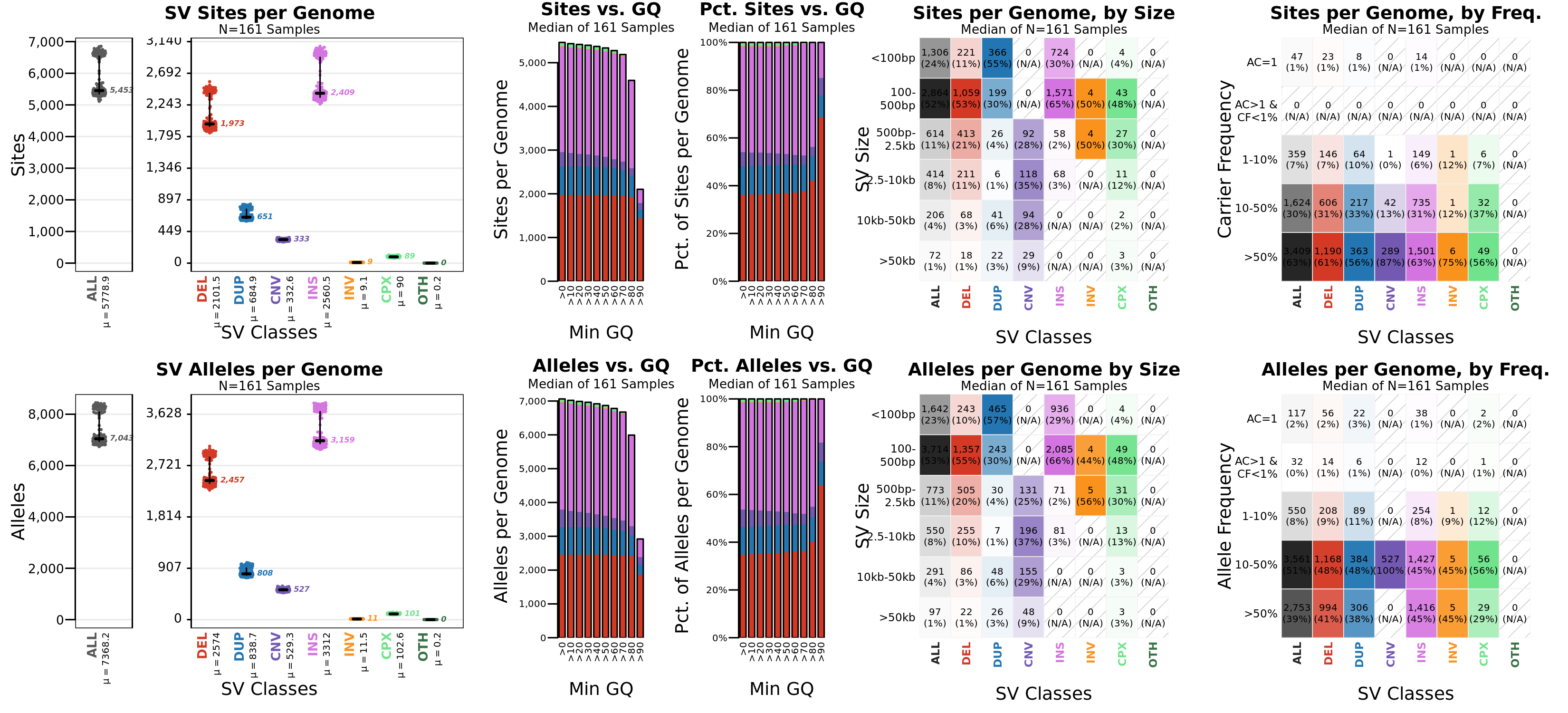
SV per genome
Shows per-genome SV count distributions broken down by SV type. The top row is a site-level analysis, and
the bottom row provides counts of individual alleles. Distributions of genotype quality (GQ), SV size, and
frequency are also shown.
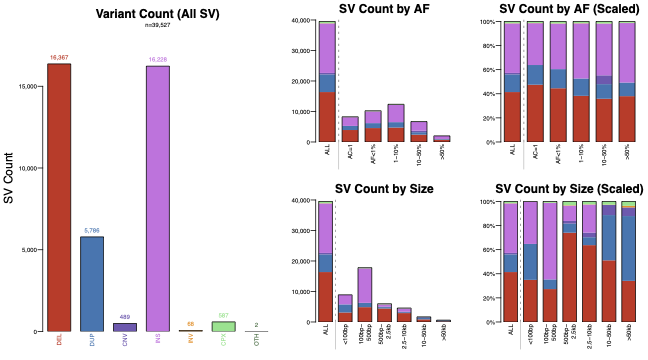
SV counts
Summarizes total variant counts by type, frequency, and size.
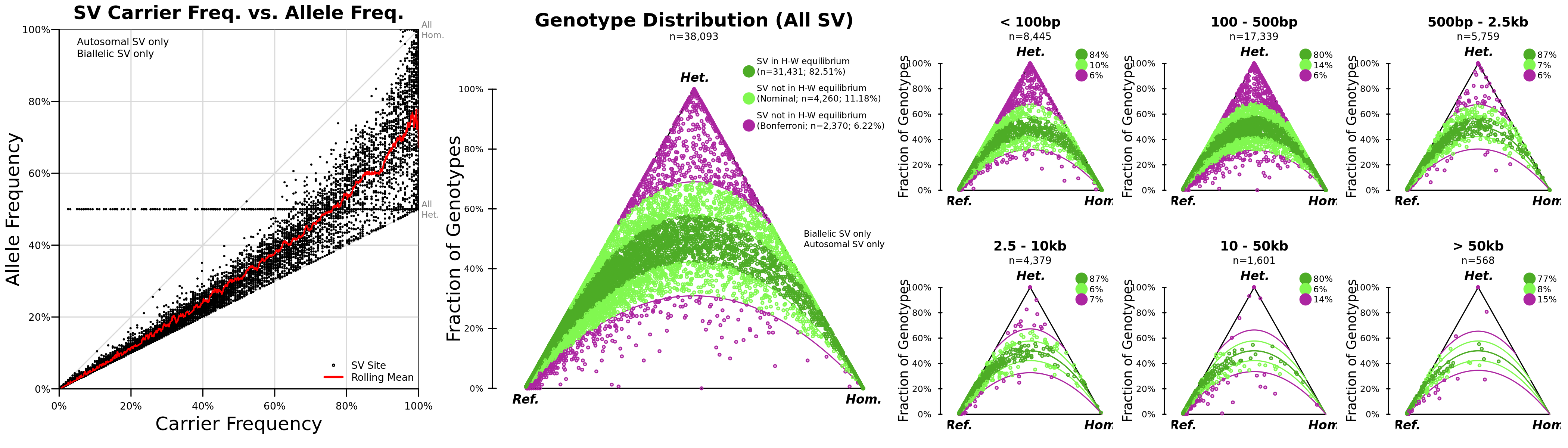
Genotype distributions
Distributions of genotypes across the call set.
The left panel plots carrier frequency against allele frequency and can be used to visualize genotyping bias. In this plot, the rolling mean should not lie on the lower or upper extremes (all het. or all hom.).
The middle panel plots the Hardy-Weinberg (HW) distribution of the cohort and provides estimates for the proportions of variants in HW equilibrium (or nominally in HW equilibrium before a multiple testing correction). The right-hand plots stratify HW by SV size.
The horizontal line bisecting the vertical axis in the AF vs. carrier frequency plot was fixed in. #625).
Size distributions
Contains plots of SV length stratified by class and frequency.
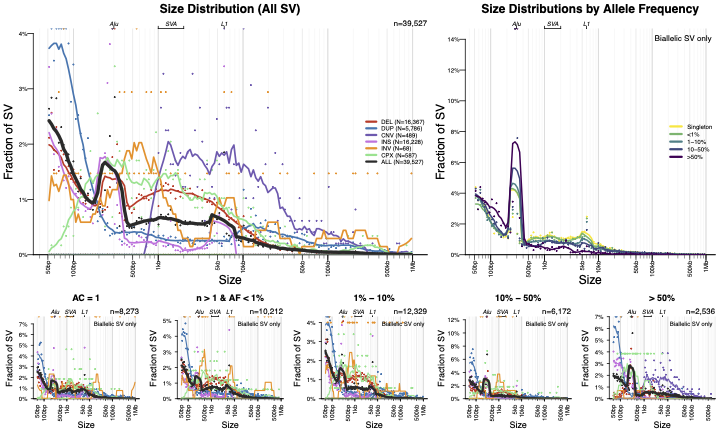
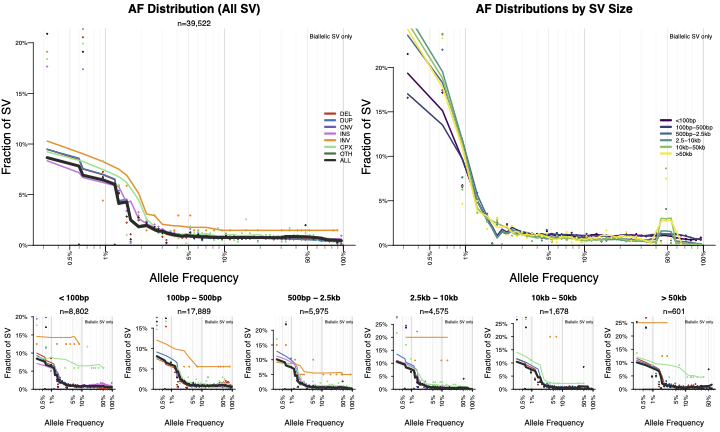
Frequency distributions
Contains plots of allele frequency stratified by class and size.
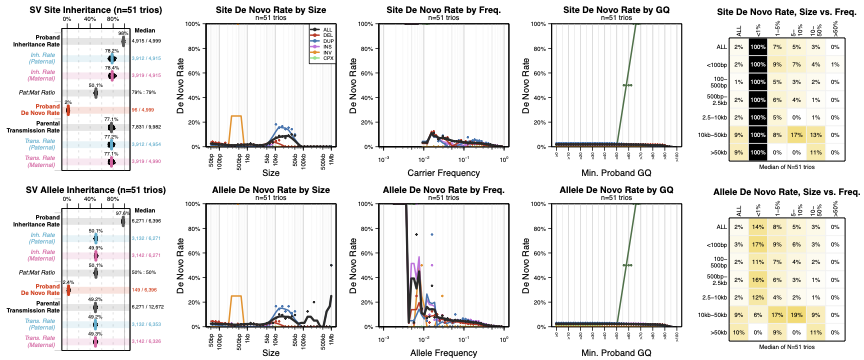
Optional SV trio inheritance
Inheritance analysis of sites and alleles, including de novo rate distributions by SV class, size, frequency, and genotype quality. This plot is only generated if the cohort contains trios defined in the PED file.
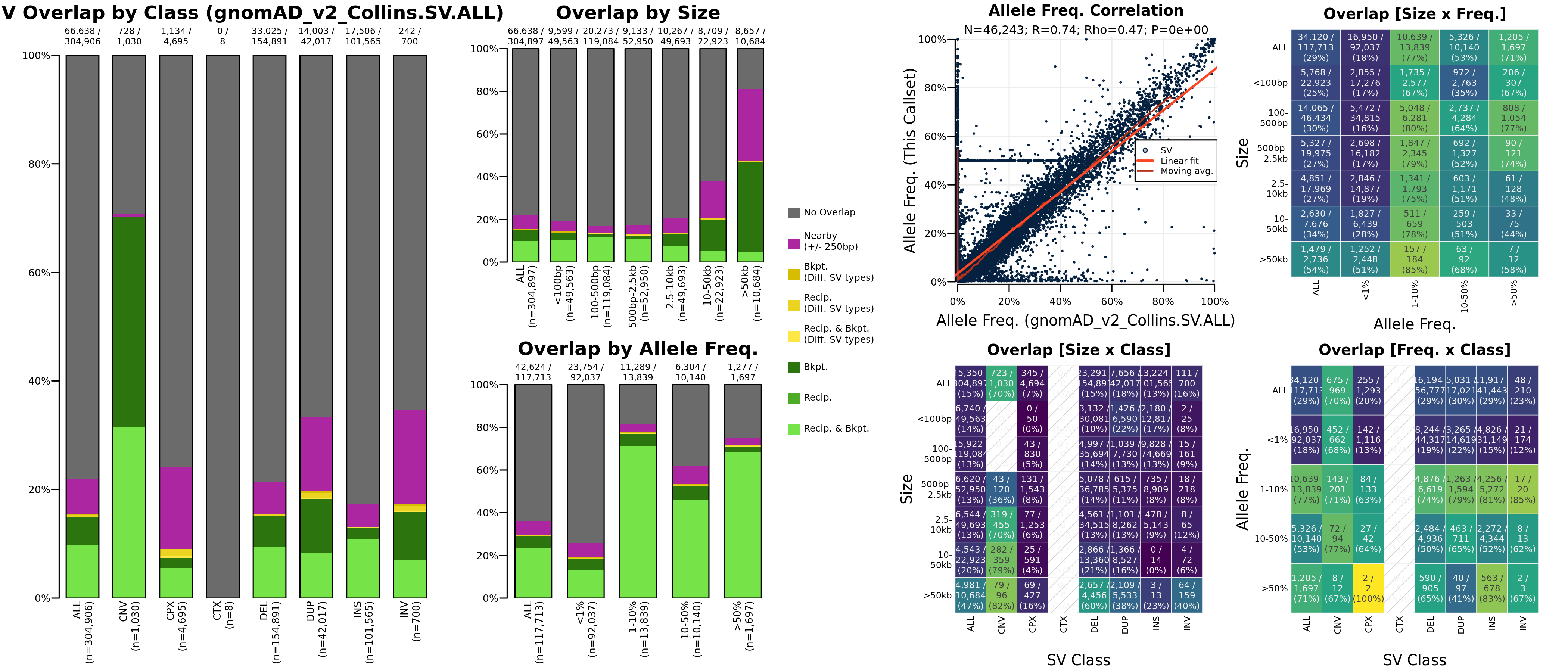
Optional Call set benchmarking
This plot will be generated for each external call set provided. The left-hand panels show distributions of overlapping variants in the input VCF by SV class, size, and frequency. Colors depict proportions of variants meeting each overlap criterion. The right-hand panels show distributions among variants overlapping between the input and external call sets by SV class, size, and frequency.
Note the horizontal line bisecting the vertical axis in the AF vs. AF plot has been fixed in #625).
Recommendations
We suggest users observe the following basic criteria to assess the overall quality of the final call set:
- Number of PASS variants per genome (which excludes
BND) between 7,000 and 11,000. - Relative overall proportions of SV types similar to those shown above.
- Similar size and frequency distributions to those shown above.
- At least 70% of variants in HW equilibrium. Note that this may be lower depending on how well the underlying cohort population structure adheres to the assumptions of the HW model. Note that HW equilibrium usually improves after genotype filtering.
- Strong allele frequency correlation against an external benchmarking dataset such as gnomAD-v2.
- Low allele de novo inheritance rate (if trios are present), typically below 10%.
These are intended as general guidelines and not strict limits on call set quality.
Inputs
vcfs
Array of one or more input VCFs. Each VCF must be indexed. This input is structured as an Array type for convenience
when VCFs are sharded by contig. All VCFs in the array are concatenated before processing.
Optional vcf_format_has_cn
Default: true. This must be set to false for VCFs from modules before CleanVcf (i.e. without CN fields).
Optional bcftools_preprocessing_options
Additional arguments to pass to bcftools view before running the VCF through QC tools.
Optional ped_file
Family structures and sex assignments determined in EvidenceQC. See PED file format. If provided and there are trios present then SV trio plots will be generated.
Optional list_of_samples_to_include
If provided, the input VCF(s) will be subset to samples in the list.
Optional sample_renaming_tsv
File with mapping to rename per-sample benchmark sample IDs for compatibility with cohort (see here for example).
Optional max_trios
Default: 1000. Upper limit on the number of trios to use for inheritance analysis.
prefix
Output prefix, such as cohort name. May be alphanumeric with underscores.
sv_per_shard
Records per shard for parallel processing. This parameter may be reduced if the workflow is running slowly.
samples_per_shard
Samples per shard for per-sample QC processing. This parameter may be reduced if the workflow is running slowly. Only has an effect when do_per_sample_qc is enabled.
Optional do_per_sample_qc
Default: true. If enabled, performs per-sample SV count plots, family analysis, and site-level benchmarking.
Optional site_level_comparison_datasets
Array of two-element arrays, one per dataset, each a tuple containing [prefix, bed_uri_path], where the latter is
a directory containing multiple bed files containing the call set stratified by subpopulation. For example:
["gnomAD_v2_Collins", "gs://gatk-sv-resources-public/hg38/v0/sv-resources/resources/v1/gnomAD_v2_Collins"]
where the bucket directory contains the following files:
gs://gatk-sv-resources-public/hg38/v0/sv-resources/resources/v1/gnomAD_v2_Collins/gnomAD_v2_Collins.SV.AFR.bed.gz
gs://gatk-sv-resources-public/hg38/v0/sv-resources/resources/v1/gnomAD_v2_Collins/gnomAD_v2_Collins.SV.ALL.bed.gz
gs://gatk-sv-resources-public/hg38/v0/sv-resources/resources/v1/gnomAD_v2_Collins/gnomAD_v2_Collins.SV.AMR.bed.gz
gs://gatk-sv-resources-public/hg38/v0/sv-resources/resources/v1/gnomAD_v2_Collins/gnomAD_v2_Collins.SV.EAS.bed.gz
gs://gatk-sv-resources-public/hg38/v0/sv-resources/resources/v1/gnomAD_v2_Collins/gnomAD_v2_Collins.SV.EUR.bed.gz
gs://gatk-sv-resources-public/hg38/v0/sv-resources/resources/v1/gnomAD_v2_Collins/gnomAD_v2_Collins.SV.OTH.bed.gz
Users can examine the above files as an example of how to format custom benchmarking datasets.
See the *_site_level_benchmarking_dataset entries in the Resource files section for available
benchmarking call sets.
Optional sample_level_comparison_datasets
Array of two-element arrays, one per dataset, each a tuple containing [prefix, tarball_uri], where the latter is
a tarball containing sample-level benchmarking data. For example:
[["HGSV_ByrskaBishop", "gs://gatk-sv-resources-public/hg38/v0/sv-resources/resources/v1/HGSV_ByrskaBishop_GATKSV_perSample.tar.gz"]]
Users can examine the above file as an example of how to format custom benchmarking datasets.
See the *_sample_level_benchmarking_dataset entries in the Resource files
section for available benchmarking call sets.
Optional random_seed
Default: 0. Random seed for sample subsetting in external call set comparisons.
Optional max_gq
Default: 99. Max value to define range for GQ plotting. For modules prior to CombineBatches, use 999.
Optional downsample_qc_per_sample
Default: 1000. Number of samples to subset to for per-sample QC.
Outputs
sv_vcf_qc_output
Tarball of QC plots and data tables.
vcf2bed_output
Bed file containing all input variants.