Filter
Description
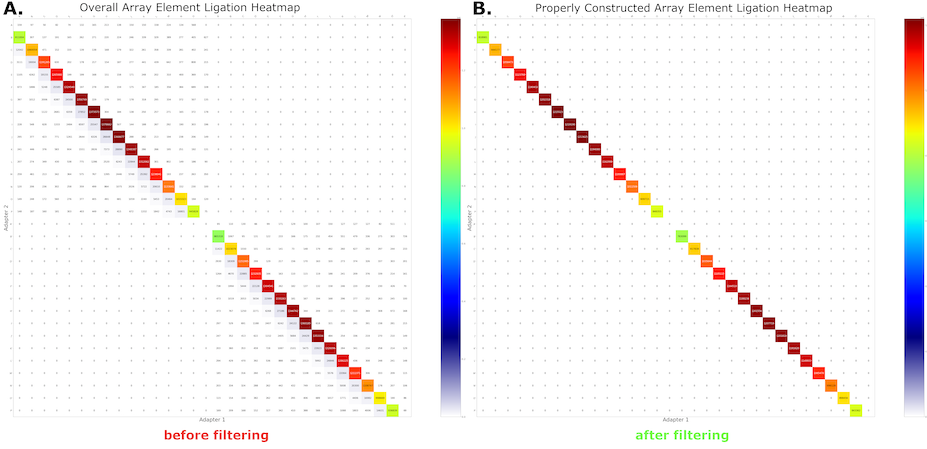
After running the annotate command on MAS-seq data, we expect that MAS-seq adapters will be found in sequential order throughout the length of the read. Reads that violate this expectation are potentially mis-segmented, and using them in downstream analysis can lead to biological misinterpretations (e.g. false fusion events, aberrant alternative splicing, erroneous transcript degradation, etc.).
Such errors manifest as off-subdiagonal elements in our ligation heatmap (left panel in figure below), depicting MAS-seq adapter adjacencies found in each read. The filter command removes the off-subdiagonal reads (right panel), ensuring that only high-quality data with confident and model-consistent segmentations are propagated to downstream analysis.
Command help
$ longbow filter --help
Usage: longbow filter [OPTIONS] INPUT_BAM
Filter reads by whether they conform to expected segment order.
Options:
-v, --verbosity LVL Either CRITICAL, ERROR, WARNING, INFO or DEBUG
-p, --pbi PATH BAM .pbi index file
-o, --out-prefix TEXT Output file prefix [required]
-m, --model TEXT The model to use for annotation. If the given value
is a pre-configured model name, then that model will
be used. Otherwise, the given value will be treated
as a file name and Longbow will attempt to read in
the file and create a LibraryModel from it. Longbow
will assume the contents are the configuration of a
LibraryModel as per LibraryModel.to_json().
[default: mas15]
-f, --force Force overwrite of the output files if they exist.
[default: False]
--help Show this message and exit.
Example
$ longbow filter -o filtered annotated.bam
[INFO 2021-08-12 10:04:26 filter] Invoked via: longbow filter -o filtered annotated.bam
[INFO 2021-08-12 10:04:26 filter] Using The standard MAS-seq 15 array element model.
[INFO 2021-08-12 10:04:28 filter] Writing reads that conform to the model to: filtered_longbow_filter_passed.bam
[INFO 2021-08-12 10:04:28 filter] Writing reads that do not conform to the model to: filtered_longbow_filter_failed.bam
[INFO 2021-08-12 10:04:28 filter] Filtering according to mas15 model ordered key adapters: A, B, C, D, E, F, G, H, I, J, K, L, M, N, O, P
Progress: 8 read [00:00, 970.54 read/s]
[INFO 2021-08-12 10:04:28 filter] Done. Elapsed time: 2.35s.
[INFO 2021-08-12 10:04:28 filter] Total Reads Processed: 8
[INFO 2021-08-12 10:04:28 filter] # Reads Passing Model Filter: 8 (100.00%)
[INFO 2021-08-12 10:04:28 filter] # Reads Failing Model Filter: 0 (0.00%)
[INFO 2021-08-12 10:04:28 filter] Total # correctly ordered key adapters in passing reads: 110
[INFO 2021-08-12 10:04:28 filter] Total # correctly ordered key adapters in failing reads: 0
[INFO 2021-08-12 10:04:28 filter] Avg # correctly ordered key adapters per passing read: 13.7500 [16]
[INFO 2021-08-12 10:04:28 filter] Avg # correctly ordered key adapters per failing read: 0.0000 [16]