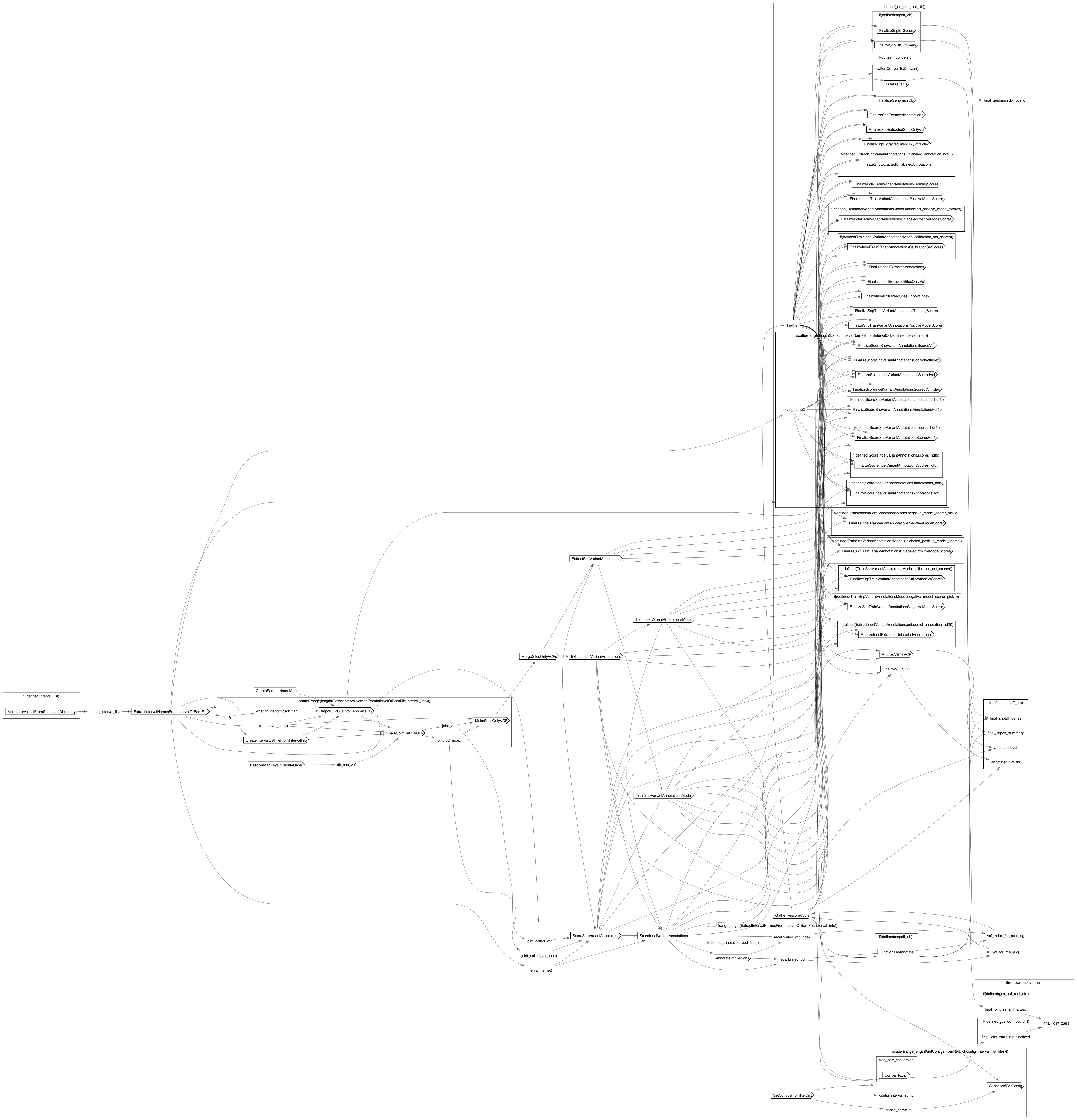
SRJointCallGVCFsWithGenomicsDBPopulationScale
SRJointCallGVCFsWithGenomicsDBPopulationScale
- author
- Jonn Smith
- description
- A workflow that performs joint calling on single-sample gVCFs from GATK4 HaplotypeCaller using GenomicsDB. This Workflow relies on previously constructed genomicsDB instances to provide population-scale context for joint calling. NOTE: Currently assumes the interval list consists of only whole contigs.
Inputs
Required
genomicsdb_tar_contig_map_file(File, required): File containing a map of contigs to GenomicsDB tar files. This file is used to determine which GenomicsDB tar file to use for each contig.gvcf_indices(Array[File], required): Array of gvcf index files forgvcfs. Order should correspond to that ingvcfs.gvcfs(Array[File], required): Array of GVCF files to use as inputs for joint calling.indel_is_calibration(Array[Boolean], required): Array of booleans indicating which files inindel_known_reference_variantsshould be used as calibration sets. True ->calibration set. False -> NOT a calibration set.indel_is_training(Array[Boolean], required): Array of booleans indicating which files inindel_known_reference_variantsshould be used as training sets. True -> training set. False -> NOT a training set.indel_known_reference_variants(Array[File], required): Array of VCF files to use as input reference variants for INDELs. Each can be designated as either calibration or training usingindel_is_trainingandindel_is_calibration.indel_known_reference_variants_identifier(Array[File], required): Array of names to give to the VCF files given inindel_known_reference_variants. Order should correspond to that inindel_known_reference_variants.indel_known_reference_variants_index(Array[File], required): Array of VCF index files forindel_known_reference_variants. Order should correspond to that inindel_known_reference_variants.prefix(String, required): Prefix to use for output files.ref_map_file(File, required): Reference map file indicating reference sequence and auxillary file locationssnp_is_calibration(Array[Boolean], required): Array of booleans indicating which files insnp_known_reference_variantsshould be used as calibration sets. True ->calibration set. False -> NOT a calibration set.snp_is_training(Array[Boolean], required): Array of booleans indicating which files insnp_known_reference_variantsshould be used as training sets. True -> training set. False -> NOT a training set.snp_known_reference_variants(Array[File], required): Array of VCF files to use as input reference variants for SNPs. Each can be designated as either calibration or training usingsnp_is_trainingandsnp_is_calibration.snp_known_reference_variants_identifier(Array[File], required): Array of names to give to the VCF files given insnp_known_reference_variants. Order should correspond to that insnp_known_reference_variants.snp_known_reference_variants_index(Array[File], required): Array of VCF index files forsnp_known_reference_variants. Order should correspond to that insnp_known_reference_variants.
Optional
annotation_bed_file_annotation_names(Array[String]?): Array of names/FILTER column entries to use for each given file inannotation_bed_files. Order should correspond toannotation_bed_files.annotation_bed_file_indexes(Array[File]?): Array of bed indexes forannotation_bed_files. Order should correspond toannotation_bed_files.annotation_bed_files(Array[File]?): Array of bed files to use to FILTER/annotate variants in the output file. Annotations will be placed in the FILTER column, effectively filtering variants that overlap these regions.background_sample_gvcf_indices(Array[Array[File]]?): Array of GVCF index files forbackground_sample_gvcfs. Order should correspond to that inbackground_sample_gvcfs.background_sample_gvcfs(Array[Array[File]]?): Array of GVCFs to use as background samples for joint calling.gcs_out_root_dir(String?): GCS Bucket into which to finalize outputs. If no bucket is given, outputs will not be finalized and instead will remain in their native execution location.interval_list(File?)snpeff_db(File?)snpeff_db_identifier(String?)AnnotateVcfRegions.runtime_attr_override(RuntimeAttr?)ConvertToZarr.ref_fai(String?)ConvertToZarr.ref_fasta(String?)CreateIntervalListFileFromIntervalInfo.runtime_attr_override(RuntimeAttr?)CreateSampleNameMap.runtime_attr_override(RuntimeAttr?)ExtractIndelVariantAnnotations.runtime_attr_override(RuntimeAttr?)ExtractIntervalNamesFromIntervalOrBamFile.runtime_attr_override(RuntimeAttr?)ExtractSnpVariantAnnotations.runtime_attr_override(RuntimeAttr?)FinalizeGenomicsDB.runtime_attr_override(RuntimeAttr?)FinalizeIndelExtractedAnnotations.name(String?)FinalizeIndelExtractedAnnotations.runtime_attr_override(RuntimeAttr?)FinalizeIndelExtractedSitesOnlyVcf.name(String?)FinalizeIndelExtractedSitesOnlyVcf.runtime_attr_override(RuntimeAttr?)FinalizeIndelExtractedSitesOnlyVcfIndex.name(String?)FinalizeIndelExtractedSitesOnlyVcfIndex.runtime_attr_override(RuntimeAttr?)FinalizeIndelExtractedUnlabeledAnnotations.name(String?)FinalizeIndelExtractedUnlabeledAnnotations.runtime_attr_override(RuntimeAttr?)FinalizeIndelTrainVariantAnnotationsCalibrationSetScores.name(String?)FinalizeIndelTrainVariantAnnotationsCalibrationSetScores.runtime_attr_override(RuntimeAttr?)FinalizeIndelTrainVariantAnnotationsNegativeModelScorer.name(String?)FinalizeIndelTrainVariantAnnotationsNegativeModelScorer.runtime_attr_override(RuntimeAttr?)FinalizeIndelTrainVariantAnnotationsPositiveModelScorer.name(String?)FinalizeIndelTrainVariantAnnotationsPositiveModelScorer.runtime_attr_override(RuntimeAttr?)FinalizeIndelTrainVariantAnnotationsTrainingScores.name(String?)FinalizeIndelTrainVariantAnnotationsTrainingScores.runtime_attr_override(RuntimeAttr?)FinalizeIndelTrainVariantAnnotationsUnlabeledPositiveModelScores.name(String?)FinalizeIndelTrainVariantAnnotationsUnlabeledPositiveModelScores.runtime_attr_override(RuntimeAttr?)FinalizeScoreIndelVariantAnnotationsAnnotationsHdf5.name(String?)FinalizeScoreIndelVariantAnnotationsAnnotationsHdf5.runtime_attr_override(RuntimeAttr?)FinalizeScoreIndelVariantAnnotationsScoredVcf.name(String?)FinalizeScoreIndelVariantAnnotationsScoredVcf.runtime_attr_override(RuntimeAttr?)FinalizeScoreIndelVariantAnnotationsScoredVcfIndex.name(String?)FinalizeScoreIndelVariantAnnotationsScoredVcfIndex.runtime_attr_override(RuntimeAttr?)FinalizeScoreIndelVariantAnnotationsScoresHdf5.name(String?)FinalizeScoreIndelVariantAnnotationsScoresHdf5.runtime_attr_override(RuntimeAttr?)FinalizeScoreSnpVariantAnnotationsAnnotationsHdf5.name(String?)FinalizeScoreSnpVariantAnnotationsAnnotationsHdf5.runtime_attr_override(RuntimeAttr?)FinalizeScoreSnpVariantAnnotationsScoredVcf.name(String?)FinalizeScoreSnpVariantAnnotationsScoredVcf.runtime_attr_override(RuntimeAttr?)FinalizeScoreSnpVariantAnnotationsScoredVcfIndex.name(String?)FinalizeScoreSnpVariantAnnotationsScoredVcfIndex.runtime_attr_override(RuntimeAttr?)FinalizeScoreSnpVariantAnnotationsScoresHdf5.name(String?)FinalizeScoreSnpVariantAnnotationsScoresHdf5.runtime_attr_override(RuntimeAttr?)FinalizeSnpEffGenes.runtime_attr_override(RuntimeAttr?)FinalizeSnpEffSummary.runtime_attr_override(RuntimeAttr?)FinalizeSnpExtractedAnnotations.name(String?)FinalizeSnpExtractedAnnotations.runtime_attr_override(RuntimeAttr?)FinalizeSnpExtractedSitesOnlyVcf.name(String?)FinalizeSnpExtractedSitesOnlyVcf.runtime_attr_override(RuntimeAttr?)FinalizeSnpExtractedSitesOnlyVcfIndex.name(String?)FinalizeSnpExtractedSitesOnlyVcfIndex.runtime_attr_override(RuntimeAttr?)FinalizeSnpExtractedUnlabeledAnnotations.name(String?)FinalizeSnpExtractedUnlabeledAnnotations.runtime_attr_override(RuntimeAttr?)FinalizeSnpTrainVariantAnnotationsCalibrationSetScores.name(String?)FinalizeSnpTrainVariantAnnotationsCalibrationSetScores.runtime_attr_override(RuntimeAttr?)FinalizeSnpTrainVariantAnnotationsNegativeModelScorer.name(String?)FinalizeSnpTrainVariantAnnotationsNegativeModelScorer.runtime_attr_override(RuntimeAttr?)FinalizeSnpTrainVariantAnnotationsPositiveModelScorer.name(String?)FinalizeSnpTrainVariantAnnotationsPositiveModelScorer.runtime_attr_override(RuntimeAttr?)FinalizeSnpTrainVariantAnnotationsTrainingScores.name(String?)FinalizeSnpTrainVariantAnnotationsTrainingScores.runtime_attr_override(RuntimeAttr?)FinalizeSnpTrainVariantAnnotationsUnlabeledPositiveModelScores.name(String?)FinalizeSnpTrainVariantAnnotationsUnlabeledPositiveModelScores.runtime_attr_override(RuntimeAttr?)FinalizeVETSTBI.name(String?)FinalizeVETSTBI.runtime_attr_override(RuntimeAttr?)FinalizeVETSVCF.name(String?)FinalizeVETSVCF.runtime_attr_override(RuntimeAttr?)FinalizeZarrs.name(String?)FinalizeZarrs.runtime_attr_override(RuntimeAttr?)FunctionallyAnnotate.runtime_attr_override(RuntimeAttr?)GatherRescoredVcfs.runtime_attr_override(RuntimeAttr?)GetContigsFromRefDict.runtime_attr_override(RuntimeAttr?)GnarlyJointCallGVCFs.input_gvcf_index(File?)GnarlyJointCallGVCFs.runtime_attr_override(RuntimeAttr?)MakeIntervalListFromSequenceDictionary.runtime_attr_override(RuntimeAttr?)MakeSitesOnlyVCF.runtime_attr_override(RuntimeAttr?)MergeSitesOnlyVCFs.runtime_attr_override(RuntimeAttr?)ScoreIndelVariantAnnotations.runtime_attr_override(RuntimeAttr?)ScoreSnpVariantAnnotations.runtime_attr_override(RuntimeAttr?)TrainIndelVariantAnnotationsModel.runtime_attr_override(RuntimeAttr?)TrainIndelVariantAnnotationsModel.unlabeled_annotation_hdf5(File?)TrainSnpVariantAnnotationsModel.runtime_attr_override(RuntimeAttr?)TrainSnpVariantAnnotationsModel.unlabeled_annotation_hdf5(File?)
Defaults
do_zarr_conversion(Boolean, default=false)heterozygosity(Float, default=0.001): Joint Genotyping Parameter - Heterozygosity value used to compute prior likelihoods for any locus. See the GATKDocs for full details on the meaning of this population genetics conceptheterozygosity_stdev(Float, default=0.01): Joint Genotyping Parameter - Standard deviation of heterozygosity for SNP and indel calling.indel_calibration_sensitivity(Float, default=0.99): VETS (ScoreVariantAnnotations) parameter - score below which INDEL variants will be filtered.indel_heterozygosity(Float, default=0.000125): Joint Genotyping Parameter - Heterozygosity for indel calling. See the GATKDocs for heterozygosity for full details on the meaning of this population genetics conceptindel_max_unlabeled_variants(Int, default=0): VETS (ExtractVariantAnnotations) parameter - maximum number of unlabeled INDEL variants/alleles to randomly sample with reservoir sampling. If nonzero, annotations will also be extracted from unlabeled sites.indel_recalibration_annotation_values(Array[String], default=["BaseQRankSum", "ExcessHet", "FS", "MQ", "MQRankSum", "QD", "ReadPosRankSum", "SOR", "DP"]): VETS (ScoreSnpVariantAnnotations/ScoreVariantAnnotations) parameter - Array of annotation names to use to create the INDEL variant scoring model and over which to score INDEL variants.shard_max_interval_size_bp(Int, default=999999999): Maximum size of the interval on each shard. This along with the given sequence dictionary determines how many shards there will be. To shard by contig, set to a very high number. Default is 999999999.snp_calibration_sensitivity(Float, default=0.99): VETS (ScoreVariantAnnotations) parameter - score below which SNP variants will be filtered.snp_max_unlabeled_variants(Int, default=0): VETS (ExtractVariantAnnotations) parameter - maximum number of unlabeled SNP variants/alleles to randomly sample with reservoir sampling. If nonzero, annotations will also be extracted from unlabeled sites.snp_recalibration_annotation_values(Array[String], default=["BaseQRankSum", "ExcessHet", "FS", "MQ", "MQRankSum", "QD", "ReadPosRankSum", "SOR", "DP"]): VETS (ScoreSnpVariantAnnotations/ScoreVariantAnnotations) parameter - Array of annotation names to use to create the SNP variant scoring model and over which to score SNP variants.ConvertToZarr.num_cpus(Int, default=4)ConvertToZarr.reference(String, default="GRCh38")GnarlyJointCallGVCFs.keep_combined_raw_annotations(Boolean, default=false)ImportGVCFsIntoGenomicsDB.extra_mem_gb(Int, default=0)MakeIntervalListFromSequenceDictionary.ignore_contigs(Array[String], default=['random', 'chrUn', 'decoy', 'alt', 'HLA', 'EBV'])TrainIndelVariantAnnotationsModel.calibration_sensitivity_threshold(Float, default=0.95)TrainSnpVariantAnnotationsModel.calibration_sensitivity_threshold(Float, default=0.95)
Outputs
vcfs_per_contig(Array[File])vcf_indices_per_contig(Array[File])genomicsDB(Array[String])joint_zarrs(Array[File]?)snpEff_summary(Array[String]?)snpEff_genes(Array[String]?)
Dot Diagram