A common first analysis for morphological datasets is the activity of the cells’ phenotypes. We will use the copairs package, which makes use of mean average precision to obtain a metric of replicability for any set of morphological profiles. In other words, it indicates how similar a given set of compounds are, relative to their negative controls, which is usually cells that have experienced no perturbation.
import polars as plimport polars.selectors as csimport seaborn as snsfrom broad_babel.query import get_mapperfrom copairs.mapimport average_precision
Downloading data from 'https://zenodo.org/records/12211976/files/babel.db' to file '/home/runner/.cache/pooch/2eaa6a2f4915f72d7100683f53982ed8-babel.db'.
We will be using the CRISPR dataset specificed in our index csv, but we will select a subset of perturbations and the controls present.
Sample perturbations and add known negative control.
jcp_ids = ( profiles.select(pl.col("Metadata_JCP2022")).unique().collect().to_series().sort())subsample = jcp_ids.sample(10, seed=42)subsample = (*subsample, "JCP2022_800002") # Add the only control in CRISPR dataprofiles_subset = profiles.filter(pl.col("Metadata_JCP2022").is_in(subsample)).collect()unique_plates = profiles_subset.filter(pl.col("Metadata_JCP2022") != subsample[-1])["Metadata_Plate"].unique()perts_controls = profiles_subset.filter(pl.col("Metadata_Plate").is_in(unique_plates))with pl.Config() as cfg: cfg.set_tbl_cols(7) # Limit the number of columns printedprint(perts_controls.head())
Finally we use the parameters from . See the copairs wiki for more details on the parameters that copairs requires.
pos_sameby = ["Metadata_JCP2022"] # We want to match perturbationspos_diffby = []neg_sameby = []neg_diffby = ["pert_type"]batch_size =20000metadata_selector = cs.starts_with(("Metadata", "pert_type"))meta = perts_controls_annotated.select(metadata_selector)features = perts_controls_annotated.select(~metadata_selector)result = average_precision( meta.to_pandas(), features.to_numpy(), pos_sameby, pos_diffby, neg_sameby, neg_diffby, batch_size,)result = pl.DataFrame( result) # We convert back to polars because we prefer how it prints dataframesresult.head()
shape: (5, 8)
Metadata_Source
Metadata_Plate
Metadata_Well
Metadata_JCP2022
pert_type
n_pos_pairs
n_total_pairs
average_precision
str
str
str
str
str
i64
i64
f64
"source_13"
"CP-CC9-R1-05"
"I23"
"JCP2022_800002"
"negcon"
419
471
0.920554
"source_13"
"CP-CC9-R1-05"
"J02"
"JCP2022_800002"
"negcon"
419
471
0.920515
"source_13"
"CP-CC9-R1-05"
"L23"
"JCP2022_800002"
"negcon"
419
471
0.931227
"source_13"
"CP-CC9-R1-05"
"O23"
"JCP2022_800002"
"negcon"
419
471
0.92076
"source_13"
"CP-CC9-R1-05"
"M02"
"JCP2022_800002"
"negcon"
419
471
0.951237
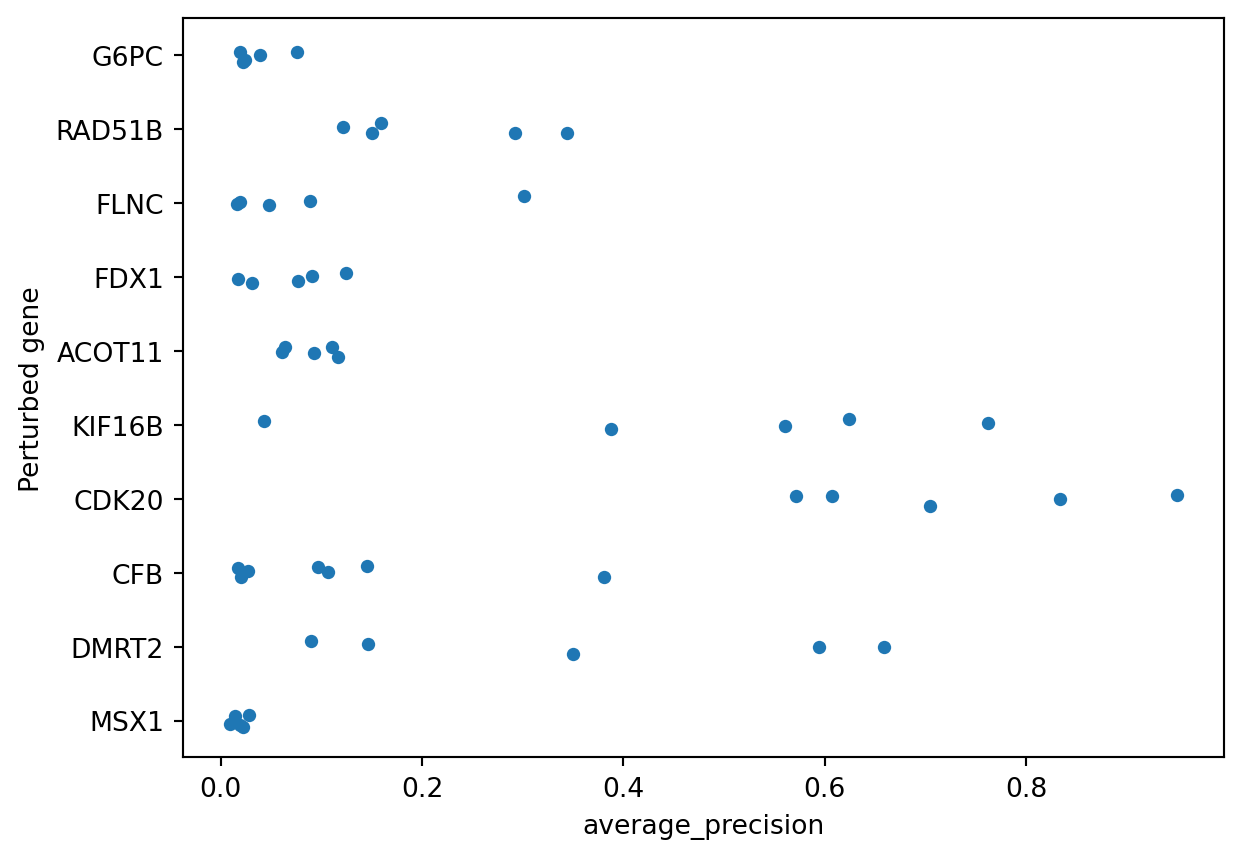
The result of copairs is a dataframe containing, in addition to the original metadata, the average precision with which perturbations were retrieved. Perturbations that look more similar to each other than to the negative controls in the plates present in the same plates will be higher. Perturbations that do not differentiate themselves against negative controls will be closer to zero.
To wrap up we pull the standard gene symbol and plot the distribution of average precision.
We can see that only some perturbations can be easily retrieved when compared to negative controls, in this case KIF16B and CDK20. For a deeper dive into how mean Average Precision (mAP) works, you can explore this notebook.