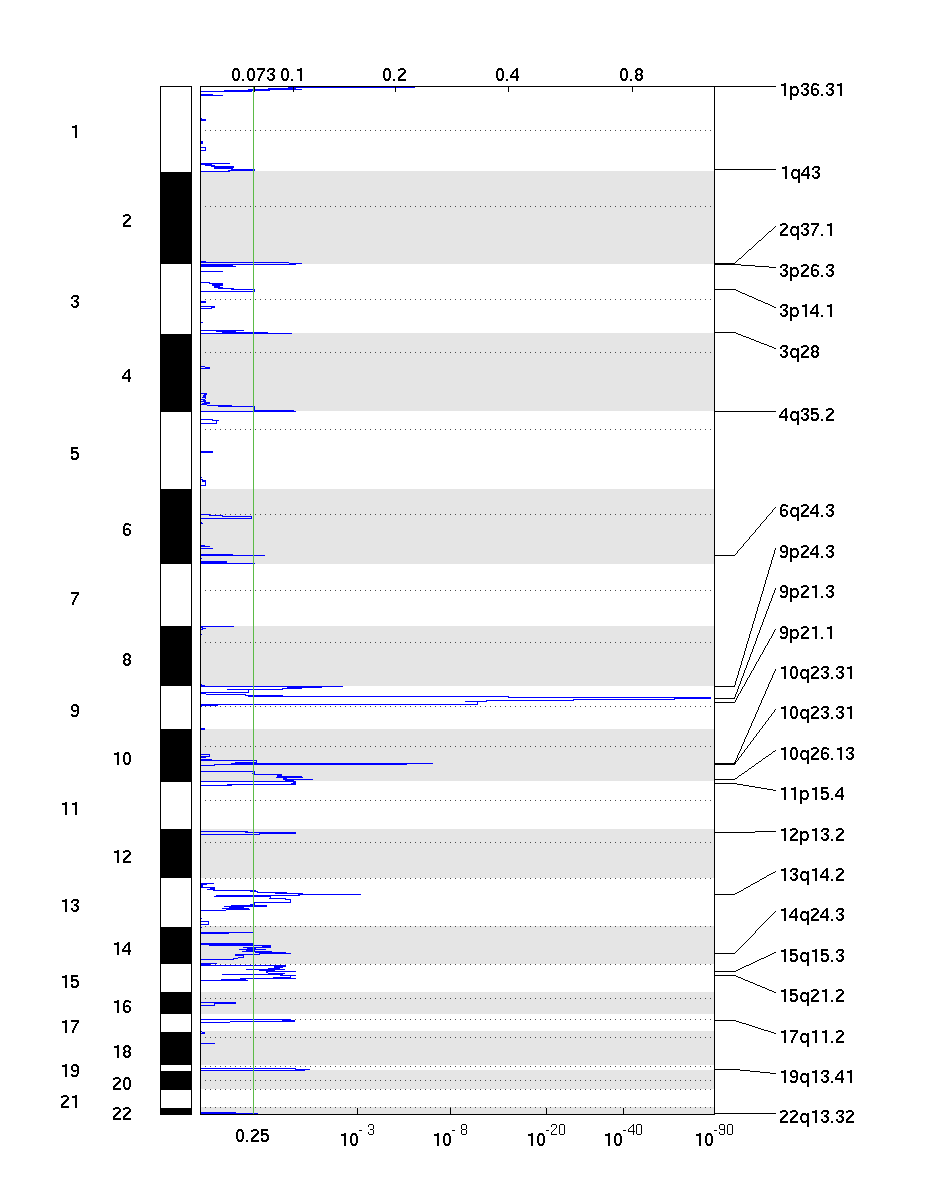
The all lesions file summarizes the results from the GISTIC run. It contains data about the significant regions of amplification and deletion as well as which samples are amplified or deleted in each of these regions. The identified regions are listed down the first column, and the samples are listed across the first row, starting in column 10.
Region Data
Columns 1-9 present the data about the significant regions as follows:
- (1) Unique Name: A name assigned to identify the region
- (2) Descriptor: The genomic descriptor of that region.
- (3) Wide Peak Limits: The "wide peak" boundaries most likely to contain the targeted genes. These are listed in genomic coordinates and marker (or probe) indices.
- (4) Peak Limits: The boundaries of the region of maximal amplification or deletion.
- (5) Region Limits: The boundaries of the entire significant region of amplification or deletion.
- (6) q-values: The q-value of the peak region.
- (7) Residual q-values: The q-value of the peak region after removing ("peeling off") amplifications or deletions that overlap other, more significant peak regions in the same chromosome.
- (8) Broad or Focal: Identifies whether the region reaches significance due primarily to broad events (called "broad"), focal events (called "focal"), or independently significant broad and focal events (called "both").
- (9) Amplitude Threshold: Key giving the meaning of values in the subsequent columns associated with each sample.
Sample Data
Each of the analyzed samples is represented in one of the columns following the lesion data (columns 10 through end). The data contained in these columns varies slightly by section of the file.
The first section can be identified by the key given in column 9 – it starts in row 2 and continues until the row that reads "Actual Copy Change Given." This section contains summarized data for each sample. A '0' indicates that the copy number of the sample was not amplified or deleted beyond the threshold amount in that peak region. A '1' indicates that the sample had low-level copy number aberrations (exceeding the low threshold indicated in column 9), and a '2' indicates that the sample had high-level copy number aberrations (exceeding the high threshold indicated in column 9).
The second section can be identified the rows in which column 9 reads "Actual Copy Change Given." The second section exactly reproduces the first section, except that here the actual changes in copy number are provided rather than zeroes, ones, and twos.
The final section is similar to the first section, except that here only broad events are included. A 1 in the samples columns (columns 10+) indicates that the median copy number of the sample across the entire significant region exceeded the threshold given in column 9. That is, it indicates whether the sample had a geographically extended event, rather than a focal amplification or deletion covering little more than the peak region.
The amp genes file contains one column for each amplification peak identified in the GISTIC analysis. The first four rows are:
- (1) cytoband
- (2) q-value
- (3) residual q-value
- (4) wide peak boundaries
These rows identify the lesion in the same way as the all lesions file.
The remaining rows list the genes contained in each wide peak. For peaks that contain no genes, the nearest gene is listed in brackets.
The del genes file contains one column for each deletion peak identified in the GISTIC analysis. The file format for the del genes file is identical to the format for the amp genes file.
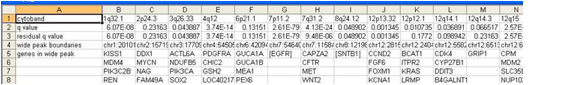
The scores file lists the q-values [presented as -log10(q)], G-scores, average amplitudes among aberrant samples, and frequency of aberration, across the genome for both amplifications and deletions. The scores file is viewable with the Genepattern SNPViewer module and may be imported into the Integrated Genomics Viewer (IGV).
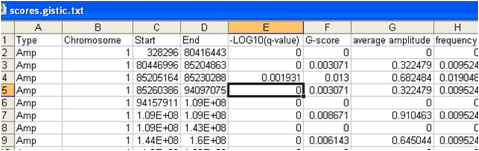
A .pdf or .png file containing a heatmap image of the genomic profiles of the segmented input copy number data. The genome is represented along the vertical axis and samples are arranged horizontally.
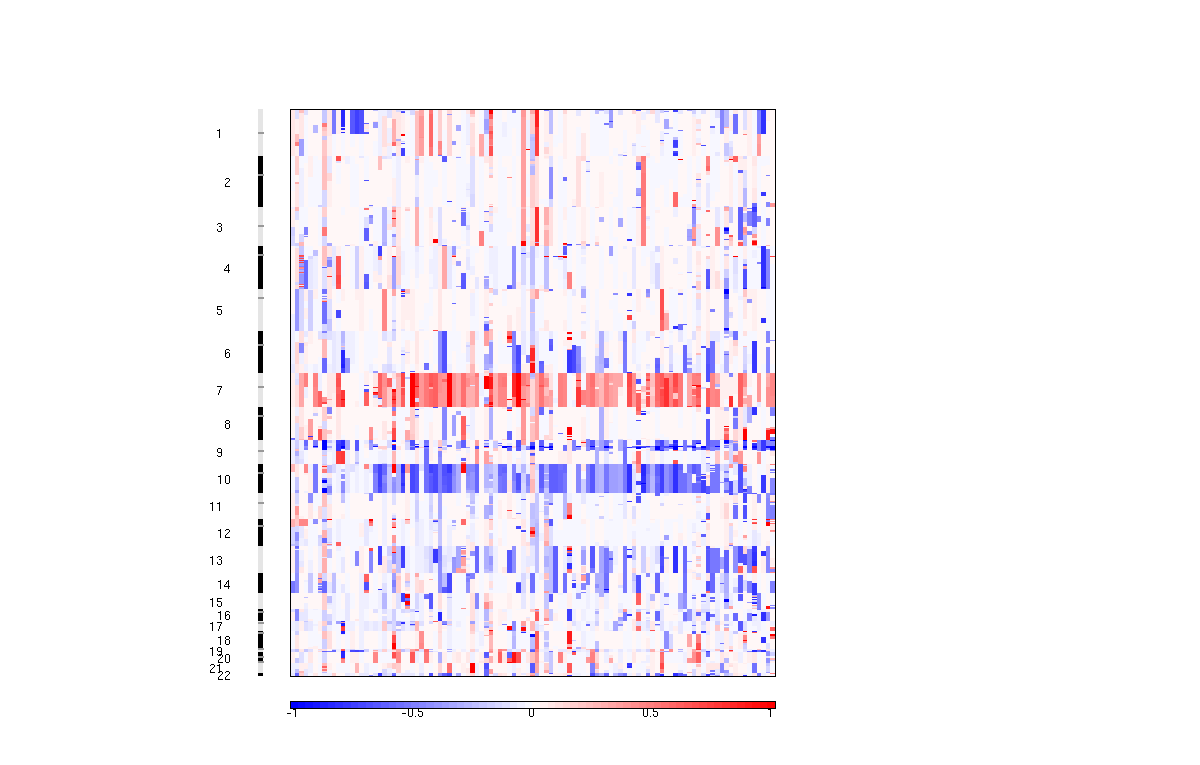
The amplification pdf is a plot of the G-scores (top) and q-values (bottom) with respect to amplifications for all markers over the entire region analyzed.
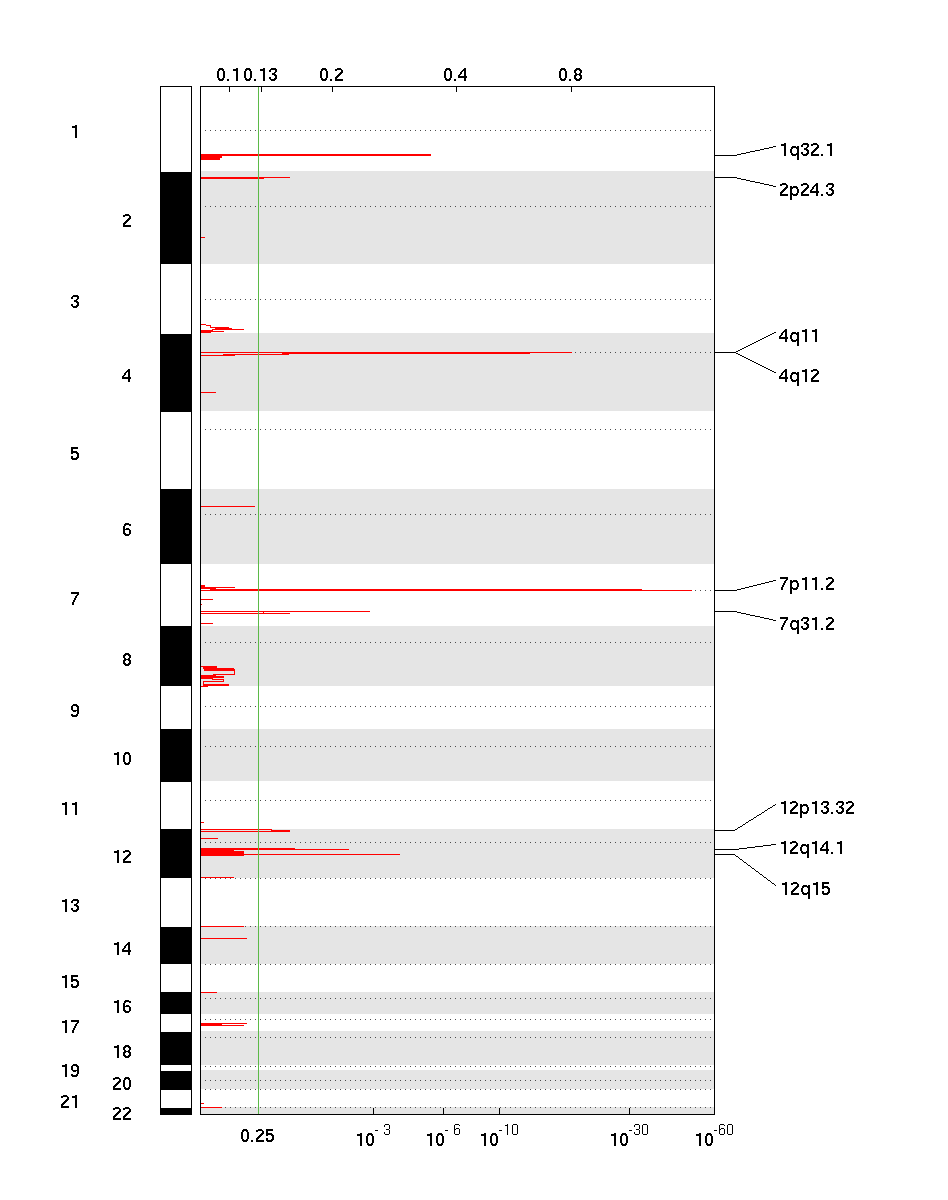
The deletion pdf is a plot of the G-scores (top) and q-values (bottom) with respect to deletions for all markers over the entire region analyzed.